This web page was produced as an assignment for Genetics 564 at UW-Madison in Spring 2014.
Conclusions
Introduction
Zellweger Syndrome Spectrum, while a collection of pretty rare diseases, has serious effects on patients and their families. There's no known cure for these diseases, and treatments are aimed solely at alleviating symptoms. Since the most common genetic cause of ZSS makes a misfolded PEX1 protein product, the primary goal of the specific aims presented here is to understand how phosphorylation affects PEX1 structure and function [1, 2]. Hopefully, by better understanding this, more translational research-based approaches can use this information to improve the folding and function of mutant PEX1.
Previous studies have shown that PEX1 interacts with PEX6 via two AAA ATPase domains to recycle PEX5, which is responsible for the transport of peroxisomal proteins that contain peroxisomal targeting sequences (PTSs) [3, 4]. This interaction is disrupted when PEX1 doesn't fold properly, and this is responsible for phenotypes seen in these mutants. Fly and mouse models have been previously established and re-capitulate phenotypes seen in humans [5, 6, 7]. Since folding efficiency is disrupted in a majority of patients, the overall objective is to investigate the potential role of phosphorylation in proper PEX1 folding. This will be studied using phosphoproteomics and by applying findings to fly and mouse models. I hypothesized that this phosphorylation affects tertiary structure and function of PEX1.
Aim 1
The main purpose of this first aim was to identify predicted and conserved phosphorylation sites in PEX1. To identify these sites, I used NetPhos 2.0, a bioinformatic tool that predicts phosphorylation sites on proteins. The algorithm scans through the amino acid sequence and assigns scores to each phosphorylatable residue (serine, threonine, tyrosine), between zero and one. The larger the number, the more likely it is that the residue is phosphorylated. Below is the data output I obtained from this algorithm for human PEX1:
Zellweger Syndrome Spectrum, while a collection of pretty rare diseases, has serious effects on patients and their families. There's no known cure for these diseases, and treatments are aimed solely at alleviating symptoms. Since the most common genetic cause of ZSS makes a misfolded PEX1 protein product, the primary goal of the specific aims presented here is to understand how phosphorylation affects PEX1 structure and function [1, 2]. Hopefully, by better understanding this, more translational research-based approaches can use this information to improve the folding and function of mutant PEX1.
Previous studies have shown that PEX1 interacts with PEX6 via two AAA ATPase domains to recycle PEX5, which is responsible for the transport of peroxisomal proteins that contain peroxisomal targeting sequences (PTSs) [3, 4]. This interaction is disrupted when PEX1 doesn't fold properly, and this is responsible for phenotypes seen in these mutants. Fly and mouse models have been previously established and re-capitulate phenotypes seen in humans [5, 6, 7]. Since folding efficiency is disrupted in a majority of patients, the overall objective is to investigate the potential role of phosphorylation in proper PEX1 folding. This will be studied using phosphoproteomics and by applying findings to fly and mouse models. I hypothesized that this phosphorylation affects tertiary structure and function of PEX1.
Aim 1
The main purpose of this first aim was to identify predicted and conserved phosphorylation sites in PEX1. To identify these sites, I used NetPhos 2.0, a bioinformatic tool that predicts phosphorylation sites on proteins. The algorithm scans through the amino acid sequence and assigns scores to each phosphorylatable residue (serine, threonine, tyrosine), between zero and one. The larger the number, the more likely it is that the residue is phosphorylated. Below is the data output I obtained from this algorithm for human PEX1:
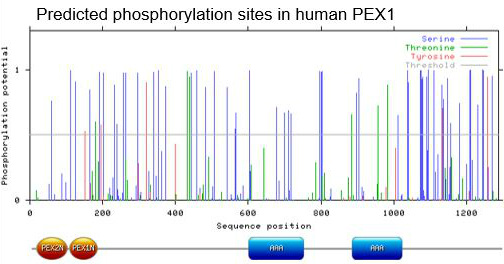
Phosphorylation potential scores for human PEX1 (NetPhos 2.0).
Since human PEX1 has a lot of potential phosphorylation sites, I focused solely on sites with a score greater than 0.5.
For the second portion of my aim, I proceeded to use the same bioinformatic tool to predict phosphorylation sites in other model organisms (B. taurus, R. norvegicus, M. musculus, D. rerio, D. melanogaster, C. elegans, A. thaliana, S. cerevisiae). You can download the raw text file of this data below:
For the second portion of my aim, I proceeded to use the same bioinformatic tool to predict phosphorylation sites in other model organisms (B. taurus, R. norvegicus, M. musculus, D. rerio, D. melanogaster, C. elegans, A. thaliana, S. cerevisiae). You can download the raw text file of this data below:
| netphos2phosphopredictions.txt |
The purpose of doing this was to compare phosphorylation sites across different organisms. The rationale for this was that if a phosphorylation site was conserved across multiple models, then it is probably important for proper function. Using Clustal Omega, I aligned these protein sequences to identify phosphorylation sites with the best conservation and highest potential scores. I narrowed the number of candidate sites down to 18, and below, they're mapped to the secondary structure of PEX1:

Well-conserved phosphorylation sites on human PEX1 (MyDomain Builder).
Chances are, these sites are the best candidates for in vivo phosphorylation, and the following two aims focus on studying the potential biological significance of these sites.
Aim 2
Since the phosphorylation sites from Aim #1 are based on algorithm predictions, it would be necessary to determine which sites are actually phosphorylated in vivo. This would help narrow down which individual sites should be studied in models. I would make use of tandem mass spectrometry to identify these sites.
Aim 2
Since the phosphorylation sites from Aim #1 are based on algorithm predictions, it would be necessary to determine which sites are actually phosphorylated in vivo. This would help narrow down which individual sites should be studied in models. I would make use of tandem mass spectrometry to identify these sites.

Tandem mass spectrometry workflow for Aim 2.
I would use reductive methylation to differentially label proteins as either a heavy isotope or light isotope. Using this, I could compare protein from wild type cells and cells with a particular amino acid change that prevents phosphorylation at a specific residue. For example, I would substitute a serine with a glycine, blocking phosphorylation at that site.
There are two outcomes I would expect from this procedure. If the mutant I've generated blocks phosphorylation, the mutant PEX1 protein would have a lower mass than wild type, because it lacks that phosphate group. However, if I've mutated a site that is not normally phosphorylated, then the masses of both the mutant and wild type PEX1 protein would be approximately the same.
The results from these experiments would help determine which sites should be studied in a model organism (Aim #3).
Aim 3
After identifying the sites where PEX1 is phosphorylated in vivo, my next step would be to determine which of these sites are required for proper PEX1 folding and function in peroxisome biogenesis. I would make use of both D. melanogaster and M. musculus models because previous literature has already established models of ZSS in these organisms.
I would generate mutants that block phosphorylation at these candidate sites, observe their phenotypes, and compare these to published literature. The rationale for this is that if any of these mutants I've generated has similar phenotypes to PEX1 loss of function mutants, then the site I've affected is crucial for proper function. Below are some phenotypes I would expect:
There are two outcomes I would expect from this procedure. If the mutant I've generated blocks phosphorylation, the mutant PEX1 protein would have a lower mass than wild type, because it lacks that phosphate group. However, if I've mutated a site that is not normally phosphorylated, then the masses of both the mutant and wild type PEX1 protein would be approximately the same.
The results from these experiments would help determine which sites should be studied in a model organism (Aim #3).
Aim 3
After identifying the sites where PEX1 is phosphorylated in vivo, my next step would be to determine which of these sites are required for proper PEX1 folding and function in peroxisome biogenesis. I would make use of both D. melanogaster and M. musculus models because previous literature has already established models of ZSS in these organisms.
I would generate mutants that block phosphorylation at these candidate sites, observe their phenotypes, and compare these to published literature. The rationale for this is that if any of these mutants I've generated has similar phenotypes to PEX1 loss of function mutants, then the site I've affected is crucial for proper function. Below are some phenotypes I would expect:
Both survival and growth rates are drastically reduced in PEX1 mutants. If my mutants are truly loss of function mutants, I'd expect them to show reduced survival and growth rates as well.
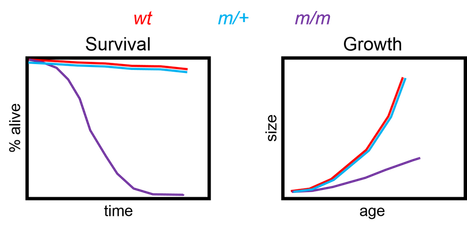
Survival and growth rates of PEX1 mutants.
If the mutants I've generated only affect PEX1 at the protein level, then I would expect PEX1 transcript levels to remain unchanged. However, the levels of PEX1 protein may be reduced. Cells have mechanisms in place to degrade misfolded proteins.
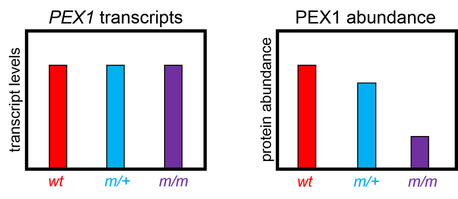
PEX1 transcript levels and PEX1 protein abundance in PEX1 mutants.
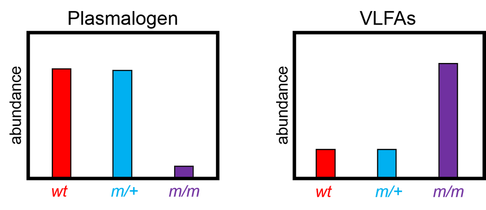
PEX1 loss of function mutants have drastically reduced levels of plasmalogens, an important component of myelin that insulates axons to preserve signal propagation in the nervous system. Peroxisomes, and therefore, PEX1, are involved in making plasmalogens, which is why mutants have less. Also cell wastes and very long chain fatty acids (VLFAs) are normally broken down by peroxisomes. This is why levels of these compounds are elevated when PEX1 is lost.
From these experiments, I would be able to identify phosphorylation sites with biological significance in PEX1 function and peroxisome biogenesis. This will give insight as to how proper PEX1 folding is achieved and how this affects its function. Eventually, this information could be used by more translational research approaches to find ways to stabilize PEX1 folding to alleviate some symptoms of ZSS patients.
Future Directions
Presumably, I will have identified at least one phosphorylation site necessary for proper PEX1 folding and function. I would first be interested in overlaying any of these phosphorylation sites with other, less characterized PEX1 mutations to see if there was any overlap.
Since there is strong evidence for the AAA ATPase domains to be important for PEX1-PEX6 interaction, and therefore, proper PEX1 function, I'd also like to investigate whether this interaction would be abolished in these phosphorylation site mutants, using co-immunoprecipitation assays. PEX1 normally binds to PEX6 to carry out its wild type function, but in a mutant affecting these AAA ATPase domains, we may find that a pulldown of PEX1 may not pull down PEX6. This would provide support that the site was affecting the proper folding and function of these AAA ATPase domains.
Since there is strong evidence for the AAA ATPase domains to be important for PEX1-PEX6 interaction, and therefore, proper PEX1 function, I'd also like to investigate whether this interaction would be abolished in these phosphorylation site mutants, using co-immunoprecipitation assays. PEX1 normally binds to PEX6 to carry out its wild type function, but in a mutant affecting these AAA ATPase domains, we may find that a pulldown of PEX1 may not pull down PEX6. This would provide support that the site was affecting the proper folding and function of these AAA ATPase domains.

Expected co-immunoprecipitation assay data pulling down wild type and mutant PEX1.
I would also be interested in seeing whether the phosphorylation sites within the N-terminal lobes of PEX1 had anything to do with proper phospholipid binding and peroxisome localization. Normally, PEX1 localizes to peroxisomes, but if a mutation affects those N-terminal lobes, PEX1 may instead be distributed evenly throughout the cell.
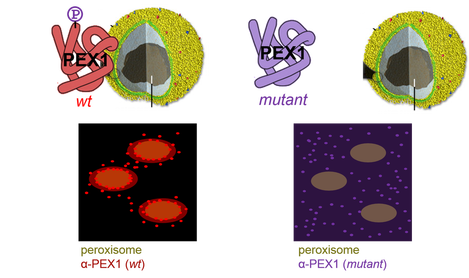
Expected cellular localization of wild type and mutant PEX1 protein.
These experiments will help add to the understanding of the phosphorylation events that occur on PEX1 and how each affects its folding and function.
| pex1_zss_final_presentation.pdf |
[1] "NINDS Zellweger Syndrome Information
Page." National Institute of
Neurological Disorders and Stroke. National Institutes of Health. U.S.
Department of Health and Human Services, 22 Oct. 2012. Web. 24 Jan. 2014. <http://www.ninds.nih.gov/disorders/zellweger/zellweger.htm>.
[2] Hans R. Waterham, Merel S. Ebberink, Genetics and molecular basis of human peroxisome biogenesis disorders, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, Volume 1822, Issue 9, September 2012, Pages 1430-1441. PMID: 22871920.
[3] Grou C, Carvalho A, Pinto M, Alencastre I, Rodrigues T, Freitas M, Francisco T, Sa-Miranda C, Azevedo J, The peroxisomal protein import machinery—a case report of transient ubiquitination with a new flavor, Cell Mol. Life Sci. 66 (2009) 254–262. PMID: 18810320.
[4] Tamura S, Matsumoto N, Imamura A, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y, Phenotype-genotype relationships in peroxisome biogenesis disorders of PEX1-defective complementation group 1 are defined by Pex1p-Pex6p interaction, Biochem J. 357 (2001) 417-426. PMID: 11439891.
[5] Hiebler S, Masuda T, Hacia J, Moser A, Faust P, Liu A, Chowdhury N, Huang N, Lauer A, Bennett J, Watkins P, Zack D, Braverman N, Raymond G, Steinberg S, The Pex1-G844D mouse: A model for mild human Zellweger spectrum disorder, Mol Genet Metab. 14 (2014) epub. PMID: 24503136.
[6] Chen H, Liu Z, Huang X, Drosophila models of peroxisomal biogenesis disorder: peroxins are required for spermatogenesis and very-long-chain fatty acid metabolism, Human Mol Genet. 19 (2010) 494-505. PMID: 19933170.
[7] Mast F, Li J, Virk M, Hughes S, Simmonds A, Rachubinski, A Drosophila model for the Zellweger spectrum of peroxisome biogenesis disorders, Dis Model Mech. 4 (2011) 659-672. PMID: 21669930 .
Image References
phosphorylation sites generated with: http://www.cbs.dtu.dk/services/NetPhos/
secondary structure generated with: http://prosite.expasy.org/mydomains/
protein: http://www.ib.bioninja.com.au/higher-level/topic-7-nucleic-acids-and/75-proteins.html
spectrometer: http://www.tcd.ie/Geology/news/articles/2012/mass-spec.php
peroxisome: http://micro.magnet.fsu.edu/cells/peroxisomes/peroxisomes.html
[2] Hans R. Waterham, Merel S. Ebberink, Genetics and molecular basis of human peroxisome biogenesis disorders, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, Volume 1822, Issue 9, September 2012, Pages 1430-1441. PMID: 22871920.
[3] Grou C, Carvalho A, Pinto M, Alencastre I, Rodrigues T, Freitas M, Francisco T, Sa-Miranda C, Azevedo J, The peroxisomal protein import machinery—a case report of transient ubiquitination with a new flavor, Cell Mol. Life Sci. 66 (2009) 254–262. PMID: 18810320.
[4] Tamura S, Matsumoto N, Imamura A, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y, Phenotype-genotype relationships in peroxisome biogenesis disorders of PEX1-defective complementation group 1 are defined by Pex1p-Pex6p interaction, Biochem J. 357 (2001) 417-426. PMID: 11439891.
[5] Hiebler S, Masuda T, Hacia J, Moser A, Faust P, Liu A, Chowdhury N, Huang N, Lauer A, Bennett J, Watkins P, Zack D, Braverman N, Raymond G, Steinberg S, The Pex1-G844D mouse: A model for mild human Zellweger spectrum disorder, Mol Genet Metab. 14 (2014) epub. PMID: 24503136.
[6] Chen H, Liu Z, Huang X, Drosophila models of peroxisomal biogenesis disorder: peroxins are required for spermatogenesis and very-long-chain fatty acid metabolism, Human Mol Genet. 19 (2010) 494-505. PMID: 19933170.
[7] Mast F, Li J, Virk M, Hughes S, Simmonds A, Rachubinski, A Drosophila model for the Zellweger spectrum of peroxisome biogenesis disorders, Dis Model Mech. 4 (2011) 659-672. PMID: 21669930 .
Image References
phosphorylation sites generated with: http://www.cbs.dtu.dk/services/NetPhos/
secondary structure generated with: http://prosite.expasy.org/mydomains/
protein: http://www.ib.bioninja.com.au/higher-level/topic-7-nucleic-acids-and/75-proteins.html
spectrometer: http://www.tcd.ie/Geology/news/articles/2012/mass-spec.php
peroxisome: http://micro.magnet.fsu.edu/cells/peroxisomes/peroxisomes.html
