This web page was produced as an assignment for Genetics 564 at UW-Madison in Spring 2014.
What is Gene Phylogeny?
Gene phylogeny allows for the examination of changes in species over time based on their gene sequences. Phylogenetic tree programs are used to construct diagrams that represent relative similarity relationships between different species.
Kalign
The following Average Distance phylogenetic tree was generated using Percent Identity (PID) method and Clustal Omega alignments. Average Distance trees are generated by putting closely related sequences at the same node with branch lengths representing estimated differences [1]. PID is calculated using the following formula:
# of equivalent aligned amino acids * 100/ smallest # of non-gaps in either sequence
This gives essentially a number of matches per 100 base pairs. In contrast, BLOSUM62 calculations simply score for base pair equivalency at each position aligned [2]. BLOSUM62 did not give a full tree with the organisms specified.
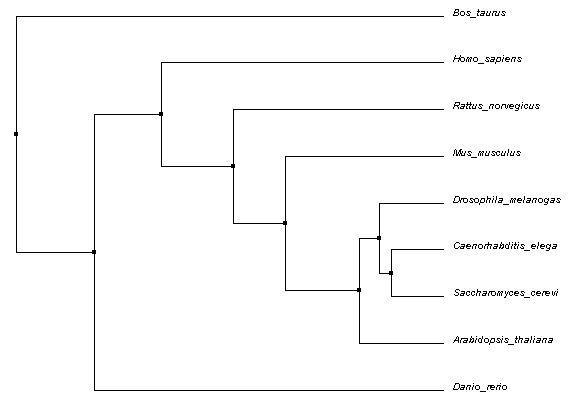
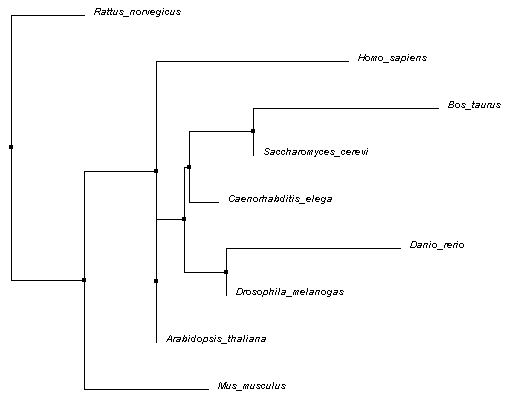
The Neighbor Joining tree shown below was also calculated using PID and Clustal Omega alignments. A Neighbor Joining tree differs from an Average Distance tree in that is will generate a tree with the shortest branches possible. Again, the BLOSUM62 tree is not shown, but can be downloaded here (Neighbor Joining).
Discussion
As discussed in the Gene Homology page, the size of the PEX1 gene limits the number of usable alignment and tree construction programs. This is why only trees derived from Kalign sequence alignment is shown.
The Average Distance tree is more plausible than the Neighbor Joining tree. The Neighbor Joining tree places two organisms from completely different phyla as being most highly related at the same node: Chordata and Ascomycota (Bos taurus and Saccharomyces cerevisiae), Chordata and Arthropoda (Danio rerio and Drosophila melanogaster), etc. It also places members of the class Mammalia at distant nodes.
The Average Distance tree is more plausible than the Neighbor Joining tree. The Neighbor Joining tree places two organisms from completely different phyla as being most highly related at the same node: Chordata and Ascomycota (Bos taurus and Saccharomyces cerevisiae), Chordata and Arthropoda (Danio rerio and Drosophila melanogaster), etc. It also places members of the class Mammalia at distant nodes.
[1] Delsuc F, Brinkmann H, Philippe H, Phylogenomics and the reconstruction of the tree of life, Nat Rev. 6 (2005) 361-375. PMID: 15861208
[2] JalView. Web. 17 Feb. 2014. <http://www.jalview.org/help/html/calculations/tree.html>.
[2] JalView. Web. 17 Feb. 2014. <http://www.jalview.org/help/html/calculations/tree.html>.

{kind=link}