This web page was produced as an assignment for Genetics 564 at UW-Madison in Spring 2014.
What is Zellweger Syndrome Spectrum?
Zellweger Syndrome Spectrum (ZSS) is a cluster of three peroxisomal biogenesis disorders (PBDs). Zellweger syndrome (ZS) is the most severe, followed by neonatal adrenoleukodystrophy (NALD), and infantile Refsum disease (IRD) [1].
A peroxisome is a cellular organelle that plays an important role in metabolic reactions. There can be between hundreds and thousands within a single cell, depending on its type [2]. They make an enzyme called catalase that uses hydrogen peroxides to oxidize and break down fatty acids and toxic waste byproducts, yielding energy for the cell. Peroxisomes are also required to make ether phospholipids and bile acids [3]. Both of these cellular roles are needed for normal development.
A peroxisome is a cellular organelle that plays an important role in metabolic reactions. There can be between hundreds and thousands within a single cell, depending on its type [2]. They make an enzyme called catalase that uses hydrogen peroxides to oxidize and break down fatty acids and toxic waste byproducts, yielding energy for the cell. Peroxisomes are also required to make ether phospholipids and bile acids [3]. Both of these cellular roles are needed for normal development.
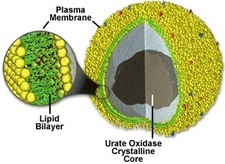 Figure 1: The peroxisome
Figure 1: The peroxisome
The lack of functional peroxisomes affects different tissues in different ways. Peroxisomes normally synthesize plasmalogens, ether phospholipids that make up myelin that insulates neurons [1]. Without myelin, signals in the brain don't travel properly [4]. Bile acids normally made in the liver aren't made properly either, affecting liver function. Fatty acids and phytanic acid build up as well.
A common strategy for studying disease involves the use of model organisms, such as yeast, worms, flies, zebrafish, mice, and rats. This allows scientists to identify genes involved in diseases and identify potential drug targets. ZSS is no different. Scientists at the University of Alberta have successfully established a fly model of ZSS and are currently using it to identify potential drug treatments:
A common strategy for studying disease involves the use of model organisms, such as yeast, worms, flies, zebrafish, mice, and rats. This allows scientists to identify genes involved in diseases and identify potential drug targets. ZSS is no different. Scientists at the University of Alberta have successfully established a fly model of ZSS and are currently using it to identify potential drug treatments:
From Youtube 2013. Zellweger syndrome discovery. Retrieved from: http://www.youtube.com/watch?v=YAQYlmNHxyc
PBDs, including ZS, are caused by mutation(s) in any of the 14 peroxin (PEX) genes. 58.9% of Zellweger Spectrum patients have a mutation in PEX1 [5].
PEX1 Gene
The human PEX1 gene has 24 exons and is located on the seventh chromosome (location 7q21.2) [accession number: NG_008341, FASTA]. It has 41,509 base pairs and 24 exons that code for a 1,283 amino acid, 143 kDa protein [accession number: NP_000457.1, FASTA].
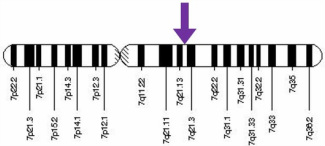
Figure 2: Human chromosome VII. The purple arrow indicates the location of the PEX1 gene (7q21.2).
PEX1 encodes a protein belonging to the AAA ATPase family (ATPases Associated with various cellular Activities) that is involved in the import of peroxisome proteins. It accomplishes this via interaction with PEX6 and anchorage to the PEX26 peroxisome membrane protein [6].
Peroxisomal proteins contain peroxisomal targeting sequences (PTSs) [7]. PEX5 recognizes and binds PTS1 [8]. PEX13 and PEX14 help dock these complexes to the peroxisome membrane. Translocation brings the proteins containing PTSs into the peroxisome. PEX2, PEX10, and PEX12 help ubiquitinate PEX5, marking it either for recycling or degradation [9]. PEX1 and PEX6 are responsible for the release of ubiquitinated PEX5 from the peroxisome membrane [6].
Peroxisomal proteins contain peroxisomal targeting sequences (PTSs) [7]. PEX5 recognizes and binds PTS1 [8]. PEX13 and PEX14 help dock these complexes to the peroxisome membrane. Translocation brings the proteins containing PTSs into the peroxisome. PEX2, PEX10, and PEX12 help ubiquitinate PEX5, marking it either for recycling or degradation [9]. PEX1 and PEX6 are responsible for the release of ubiquitinated PEX5 from the peroxisome membrane [6].
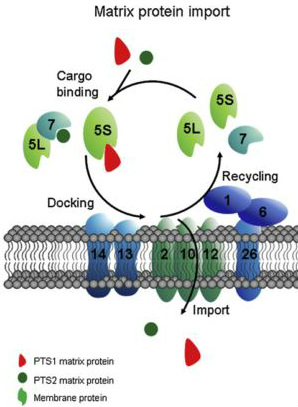
FIgure 3: Peroxisomal matrix protein import involves PEX1, PEX2, PEX5, PEX7, PEX10, PEX12, PEX13, PEX14, PEX26.
As of 2011, 90 mutations in PEX1 have been identified [10]. These mutations can lead to PBDs. A majority of these mutations are found in exons 13 and 15. The most common results in the glycine amino acid substitution with aspartic acid at location 843, within the second ATP-binding domain. This reduces PEX6 interaction [11]. Another common mutation shifts the reading frame, leading to a premature stop. This RNA product is also present at very low levels, so it's possible that the transcript is being targeted for decay [12].
Diagnosis, Screening, and Treatment
ZS symptoms are present at birth. These can include low muscle tone, cataracts and glaucoma, seizures, kidney cysts, liver malfunction, and bone abnormalities (chondrodysplasia punctate) [13]. Currently, there is no cure for ZS. Once an affected baby is born, much of the damage caused by the disorder has already been done. Treatments seek to lessen the severity of symptoms and provide comfort. Most ZS patients do not live beyond six months of age [1].
IRD and NALD often show similar symptoms to ZS, but these present themselves throughout childhood. NALD patients often do not survive through their teen years, while IRD patients have the potential to reach adulthood [14].
Diagnostic testing is often conducted using blood plasma and skin fibroblasts. Doctors test for heightened levels of very long chain fatty acids in blood plasma. These findings are often confirmed by studies of skin fibroblasts and their levels of toxic byproducts that peroxisomes are normally charged with breaking down as well as measuring catalase activity [15].
The severity of ZSS increases the importance of carrier screening and prenatal testing, especially for those who have a family history of the disorders on this spectrum.
IRD and NALD often show similar symptoms to ZS, but these present themselves throughout childhood. NALD patients often do not survive through their teen years, while IRD patients have the potential to reach adulthood [14].
Diagnostic testing is often conducted using blood plasma and skin fibroblasts. Doctors test for heightened levels of very long chain fatty acids in blood plasma. These findings are often confirmed by studies of skin fibroblasts and their levels of toxic byproducts that peroxisomes are normally charged with breaking down as well as measuring catalase activity [15].
The severity of ZSS increases the importance of carrier screening and prenatal testing, especially for those who have a family history of the disorders on this spectrum.

|

|

|
Banner image retrieved from: <http://www.iatsmartdial.com/2013/10/25/compliant-dialing-its-in-our-dna-how-about-yours>.
[1] "NINDS Zellweger Syndrome Information Page." National Institute of Neurological Disorders and Stroke. National Institutes of Health. U.S. Department of Health and Human Services, 22 Oct. 2012. Web. 24 Jan. 2014. <http://www.ninds.nih.gov/disorders/zellweger/zellweger.htm>.
[2] S.J. Huybrechts, P.P. Van Veldhoven, C. Brees, G.P. Mannaerts, G.V. Los, M. Fransen, Peroxisome dynamics in cultured mammalian cells, Traffic 10 (2009) 1722–1733. PMID: 19719477.
[3] Cooper GM. The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000. Peroxisomes. Available from: <http://www.ncbi.nlm.nih.gov/books/NBK9930>.
[4] "Myelin." National Multiple Sclerosis Society. Web. 24 Jan. 2014. <http://www.nationalmssociety.org/about-multiple-sclerosis/what-we-know-about-ms/what-is-ms/myelin/index.aspx>.
[5] Hans R. Waterham, Merel S. Ebberink, Genetics and molecular basis of human peroxisome biogenesis disorders, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, Volume 1822, Issue 9, September 2012, Pages 1430-1441. PMID: 22871920.
[6] C.P. Grou, A.F. Carvalho, M.P. Pinto, I.S. Alencastre, T.A. Rodrigues, M.O. Freitas, T. Francisco, C. Sa-Miranda, J.E. Azevedo, The peroxisomal protein import machin- ery—a case report of transient ubiquitination with a new flavor, Cell Mol. Life Sci. 66 (2009) 254–262. PMID: 18810320.
[7] Y. Elgersma, A. Vos, M. van den Berg, C.W. van Roermund, P. van der Sluijs, B. Distel, H.F. Tabak, Analysis of the carboxyl-terminal peroxisomal targeting signal 1 in a homologous context in Saccharomyces cerevisiae, J. Biol. Chem. 271 (1996) 26375–26382. PMID: 8824293.
[8] G. Dodt, D. Warren, E. Becker, P. Rehling, S.J. Gould, Domain mapping of human PEX5 reveals functional and structural similarities to Saccharomyces cerevisiae Pex18p and Pex21p, J. Biol. Chem. 276 (2001) 41769–41781. PMID: 11546814.
[9] C.C. Chang, D.S. Warren, K.A. Sacksteder, S.J. Gould, PEX12 interacts with PEX5 and PEX10 and acts downstream of receptor docking in peroxisomal matrix protein import, J. Cell Biol. 147 (1999) 761–774. PMID: 10562279.
[10] M.S. Ebberink, P.A. Mooijer, J. Gootjes, J. Koster, R.J. Wanders, H.R. Waterham, Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder, Hum. Mutat. 32 (2011) 59–69. PMID: 21031596.
[11] A. Imamura, S. Tamura, N. Shimozawa, Y. Suzuki, Z. Zhang, T. Tsukamoto, T. Orii, N. Kondo, T. Osumi, Y. Fujiki, Temperature-sensitive mutation in PEX1 moder- ates the phenotypes of peroxisome deficiency disorders, Hum. Mol. Genet. 7 (1998) 2089–2094. PMID: 9817926.
[12] C.S. Collins, S.J. Gould, Identification of a common PEX1 mutation in Zellweger syndrome, Hum. Mutat. 14 (1999) 45–53. PMID: 10447258.
[13] S. Weller, S.J. Gould, D. Valle, Peroxisome biogenesis disorders, Annu. Rev. Genomics Hum. Genet. 4 (2003) 165–211. PMID: 14527301.
[14] B.T. Poll-The, J.M. Saudubray, H.A.M. Ogier, M. Odievre, J.M. Scotto, L. Monnens, L.C.P. Govaerts, F. Roels, A. Cornelis, R.B.H. Schutgens, R.J.A. Wanders, A.W. Schram, J.M. Tager, Infantile Refsumdisease: an inherited peroxisomal disorder. Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy, Eur. J. Pediatr. 146 (1987) pp. 477–483. PMID: 2445576.
[15] R.J. Wanders, H.R. Waterham, Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders, Clin. Genet. 67 (2005) 107–133. PMID: 15679822.
Image References
peroxisome: http://micro.magnet.fsu.edu/cells/peroxisomes/peroxisomes.html
youtube video: http://www.youtube.com/watch?v=YAQYlmNHxyc
chromosomal location: http://ghr.nlm.nih.gov/gene/PEX1
peroxisome protein transport: http://www.sciencedirect.com/science/article/pii/S0925443912000932
[1] "NINDS Zellweger Syndrome Information Page." National Institute of Neurological Disorders and Stroke. National Institutes of Health. U.S. Department of Health and Human Services, 22 Oct. 2012. Web. 24 Jan. 2014. <http://www.ninds.nih.gov/disorders/zellweger/zellweger.htm>.
[2] S.J. Huybrechts, P.P. Van Veldhoven, C. Brees, G.P. Mannaerts, G.V. Los, M. Fransen, Peroxisome dynamics in cultured mammalian cells, Traffic 10 (2009) 1722–1733. PMID: 19719477.
[3] Cooper GM. The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000. Peroxisomes. Available from: <http://www.ncbi.nlm.nih.gov/books/NBK9930>.
[4] "Myelin." National Multiple Sclerosis Society. Web. 24 Jan. 2014. <http://www.nationalmssociety.org/about-multiple-sclerosis/what-we-know-about-ms/what-is-ms/myelin/index.aspx>.
[5] Hans R. Waterham, Merel S. Ebberink, Genetics and molecular basis of human peroxisome biogenesis disorders, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, Volume 1822, Issue 9, September 2012, Pages 1430-1441. PMID: 22871920.
[6] C.P. Grou, A.F. Carvalho, M.P. Pinto, I.S. Alencastre, T.A. Rodrigues, M.O. Freitas, T. Francisco, C. Sa-Miranda, J.E. Azevedo, The peroxisomal protein import machin- ery—a case report of transient ubiquitination with a new flavor, Cell Mol. Life Sci. 66 (2009) 254–262. PMID: 18810320.
[7] Y. Elgersma, A. Vos, M. van den Berg, C.W. van Roermund, P. van der Sluijs, B. Distel, H.F. Tabak, Analysis of the carboxyl-terminal peroxisomal targeting signal 1 in a homologous context in Saccharomyces cerevisiae, J. Biol. Chem. 271 (1996) 26375–26382. PMID: 8824293.
[8] G. Dodt, D. Warren, E. Becker, P. Rehling, S.J. Gould, Domain mapping of human PEX5 reveals functional and structural similarities to Saccharomyces cerevisiae Pex18p and Pex21p, J. Biol. Chem. 276 (2001) 41769–41781. PMID: 11546814.
[9] C.C. Chang, D.S. Warren, K.A. Sacksteder, S.J. Gould, PEX12 interacts with PEX5 and PEX10 and acts downstream of receptor docking in peroxisomal matrix protein import, J. Cell Biol. 147 (1999) 761–774. PMID: 10562279.
[10] M.S. Ebberink, P.A. Mooijer, J. Gootjes, J. Koster, R.J. Wanders, H.R. Waterham, Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder, Hum. Mutat. 32 (2011) 59–69. PMID: 21031596.
[11] A. Imamura, S. Tamura, N. Shimozawa, Y. Suzuki, Z. Zhang, T. Tsukamoto, T. Orii, N. Kondo, T. Osumi, Y. Fujiki, Temperature-sensitive mutation in PEX1 moder- ates the phenotypes of peroxisome deficiency disorders, Hum. Mol. Genet. 7 (1998) 2089–2094. PMID: 9817926.
[12] C.S. Collins, S.J. Gould, Identification of a common PEX1 mutation in Zellweger syndrome, Hum. Mutat. 14 (1999) 45–53. PMID: 10447258.
[13] S. Weller, S.J. Gould, D. Valle, Peroxisome biogenesis disorders, Annu. Rev. Genomics Hum. Genet. 4 (2003) 165–211. PMID: 14527301.
[14] B.T. Poll-The, J.M. Saudubray, H.A.M. Ogier, M. Odievre, J.M. Scotto, L. Monnens, L.C.P. Govaerts, F. Roels, A. Cornelis, R.B.H. Schutgens, R.J.A. Wanders, A.W. Schram, J.M. Tager, Infantile Refsumdisease: an inherited peroxisomal disorder. Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy, Eur. J. Pediatr. 146 (1987) pp. 477–483. PMID: 2445576.
[15] R.J. Wanders, H.R. Waterham, Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders, Clin. Genet. 67 (2005) 107–133. PMID: 15679822.
Image References
peroxisome: http://micro.magnet.fsu.edu/cells/peroxisomes/peroxisomes.html
youtube video: http://www.youtube.com/watch?v=YAQYlmNHxyc
chromosomal location: http://ghr.nlm.nih.gov/gene/PEX1
peroxisome protein transport: http://www.sciencedirect.com/science/article/pii/S0925443912000932
